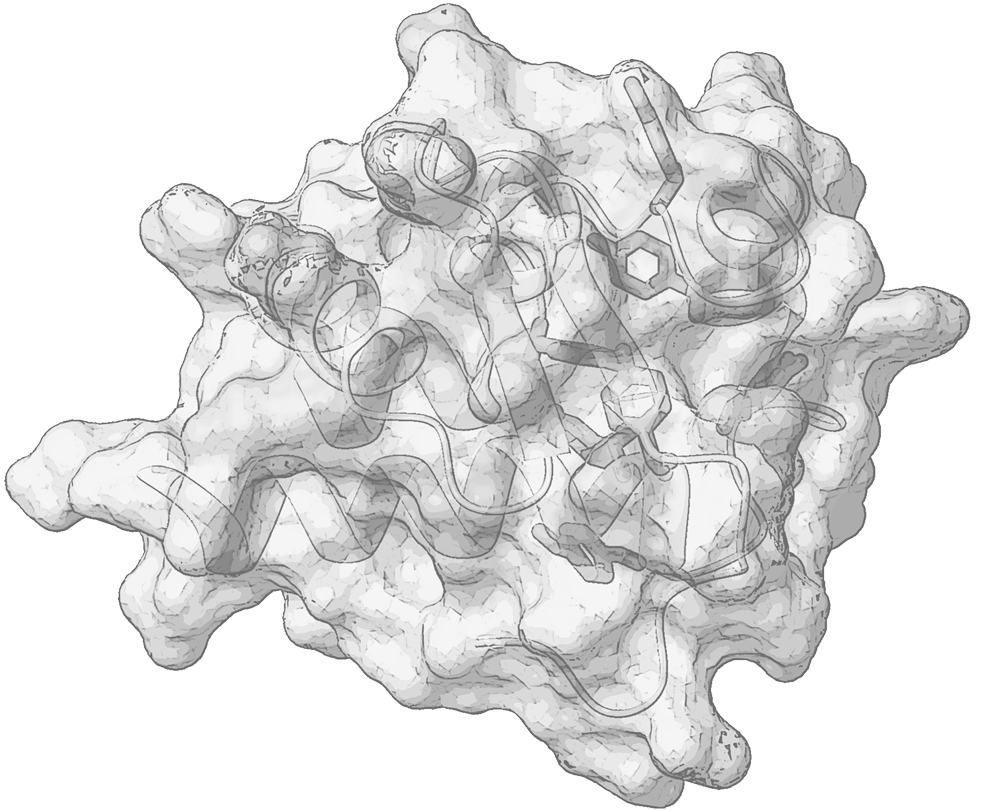
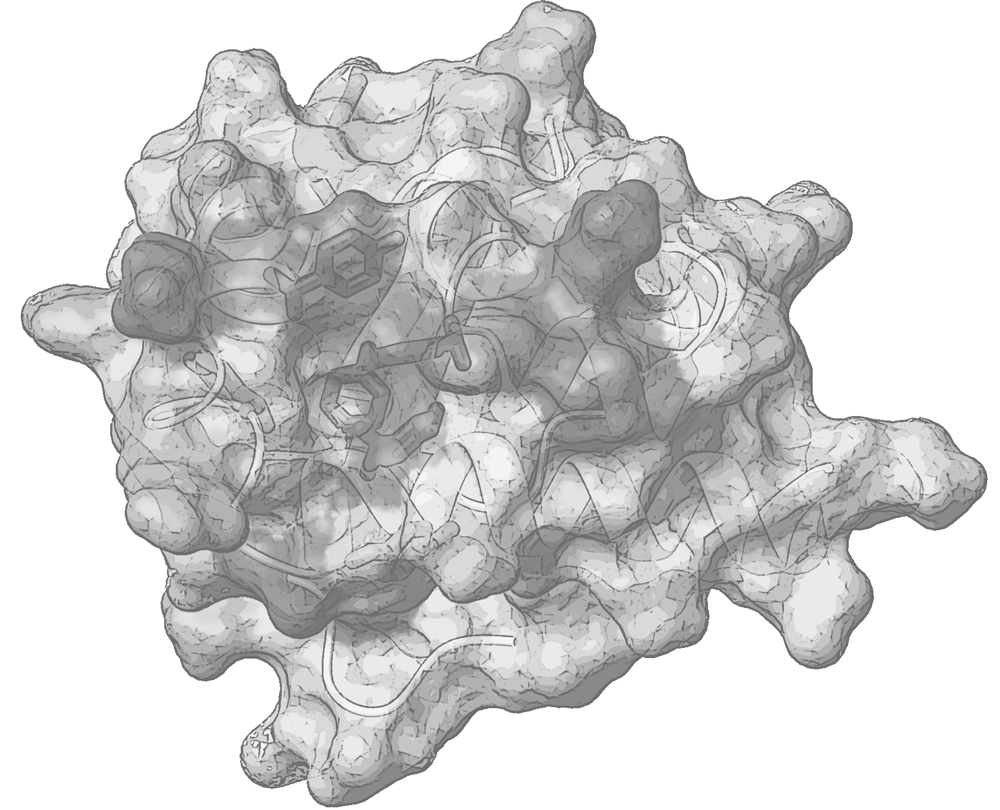
Polypeptides tend to become organized independently into a variety of functional scaffolds and to socialize with other partner molecules, forming small biomolecular complexes or large assemblies, in various intersections of biological pathways. Proteins’ architecture, plasticity and interaction determine the fate of a variety of biological events. Understanding the functional role of peptides and proteins is strongly coupled to the atomic-level insights of their structure and flexibility.
The research activities of the group encompass the study of the structure and dynamics of peptides and proteins involved in signalling pathways, proteolysis and ion/metal transfer, both in silico and in vitro.
Research Highlights
Conformational dynamics of protein and protein complexes. Our group applies NMR and other biophysical tools to study several proteins involved in different biochemical, disease-related, pathways such as (a) the ubiquitination pathway (J Mol Biol 29:2373-2386, 2017. doi: 10.1016/j.jmb.2017.06.012), (b) the NO-signalling pathway, focusing on the HNOX domain homologues of soluble Guanylate Cyclase (, (c) the ADP-ribosylation pathway, with a special interest on human and viral enzymes from SARS, MERS, SARS-CoV-2, MAYV, CHIKV, HEV1 and other viruses (Front Mol Biosci 8:653148, 2021. doi: 10.3389/fmolb.2021.653148), and (d) RNA recognition (J Mol Biol. 432:166712, 2020. doi: 10.1016/j.jmb.2020.11.011).
NMR Metabolomics. The group analyzes biological samples through NMR spectroscopy and biostatistics, to identify and quantify several metabolites as biomarkers in different families of healthy and diseased individuals, such as (a) newborns to identify metabolic disorders (Metabolomics 16:58, 2020. doi: 10.1007/s11306-020-01680-4.), (b) adults suffer bty different type of Chronic Kidney Disease (CDK) (Metabolites 12:490, 2022. doi: 10.3390/metabo12060490), (c) children with premature adrenarche (Metabolomics 18:78, 2022. doi: 10.1007/s11306-022-01941-4). Additionally, it studies the effect of the drug administration at cellular level (Cancers 13:2877, 2021. doi: 10.3390/cancers13122877).
Blue biotechnology – The MARISURF Project: Novel, Sustainable marine Bio-surfactant / Bio-emulsifiers for commercial exploitation (H2020-BG-2014-2, 635340) to analyze and characterize new eco-friendly end-products, effectively procuced in biological systems, contributing to the implementation of the objectives of the EU Blue Growth (Biomolecules 10:Ε885, 2020. doi: 10.3390/biom10060885; Microbial Cell Factories 18:164, 2019 doi: 10.1186/s12934-019-1216-8;Applied Microbiology and Biotechnology 102:8537–8549, 2018. doi: 10.1007/s00253-018-9202-3).


Research Training Opportunities
The group offer research training opportunities to Pharmacy, Chemistry, Biology or Medicine under- and post-graduate students who want to get involved in research training activities in the frame of their Diploma Thesis or to acquire a Master or a Ph.D. degree. Read more…
Students will receive training in the fields of protein expression and characterization, analysis of NMR spectra of peptides and proteins, structure determination of polypeptides using NMR structural information and Biomolecular Simulations and they will work on one of the research projects of the group.
No previous experience is required. All you need is talent and enthusiasm!
New Job & Research training opportunities for Post-Docs, & PhD candidates, through the new ERA Chair project “ESPERANCE”.
More info are found here
Hot & Fresh Papers
COVID19-NMR Consortium Comprehensive Fragment Screening of the SARS-CoV-2 Proteome Explores Novel Chemical Space for Drug Development. Angew Chem Int Ed Engl 61:e202205858, 2022. doi: 10.1002/anie.202205858.
Birkou M, Delegkou GN, Marousis KD, Fragkaki N, Toro T, Episkopou V, Spyroulias GA. Unveiling the Essential Role of Arkadia's Non-RING Elements in the Ubiquitination Process. Int J Mol Sci 23:10585, 2022. doi: 10.3390/ijms231810585.
Tsika AC, Gallo A, Fourkiotis NK, Argyriou AI, et al. Binding Adaptation of GS-441524 Diversifies Macro Domains and Downregulates SARS-CoV-2 de-MARylation Capacity. J Mol Biol 434:167720, 2022. doi: 10.1016/j.jmb.2022.167720.
Matzarapi K, Giannakopoulos A, Chasapi SA, Kritikou D, Efthymiadou A, Chrysis D, Spyroulias GA. NMR-based metabolic profiling of children with premature adrenarche. Metabolomics 18:78, 2022. doi: 10.1007/s11306-022-01941-4.